
What are Clinical Trials?
Clinical trials can be grouped into two categories: interventional and non-interventional (observational or online) trials. Interventional trials are conducted to collect data regarding the safety and efficacy of cancer therapies.
Drug testing in people requires Institutional Review Board (IRB) and Food and Drug Administration (FDA) approval in each instance. The IRB is a committee entrusted with reviewing research involving human subjects to protect the rights and welfare of research participants by assessing the safety of research proposals and ensuring the patient consent form is complete and accurate. Prior to treatment in humans, extensive laboratory research that can involve years of experiments in animals and human cells is completed. If the laboratory research is successful, researchers then request approval from the IRB and FDA to test the therapy in humans.
FDA-Approved Drugs
The FDA regulates the approval and marketing of all drugs that are sold in the United States. These drugs must be proven safe and effective by the FDA before companies can put them out on the market.
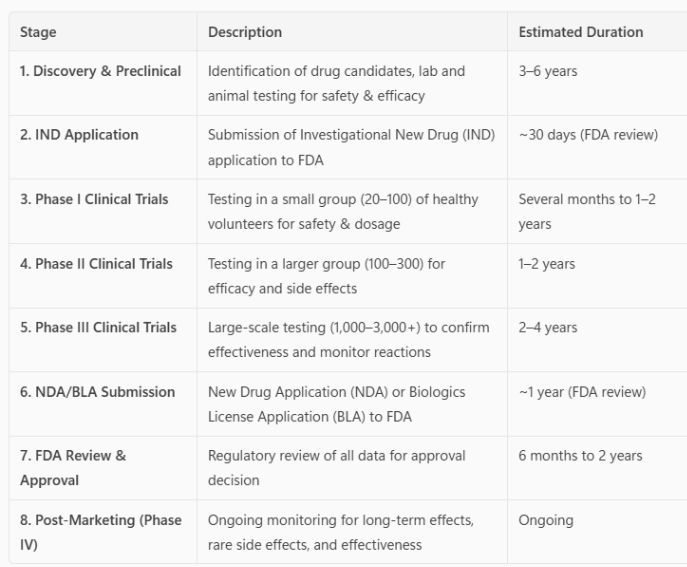
Table 1. Summary of the Drug Development Process
Human Lung Cancer Clinical Trial Phases
Phase I studies primarily assess the safety of a drug. They can be the very first time the drug has been administered to humans (called first-in-human (FIH) studies), or it can be an FDA-approved drug that is being tested in a different type of cancer or is an FDA-approved drug in combination with another drug(s). This initial phase of testing can last for several years because the trial design slowly increases the drug dose (called dose escalation) to identify the side effects before they are severe. The study will also determine how the drug(s) are absorbed, metabolized, and excreted.
Phase II studies primarily assess the efficacy of a drug. This second phase of testing can last a year or more. Some phase II studies are randomized trials where one group of patients receives the experimental drug while a second “control” group receives a standard treatment. This allows investigators to provide the pharmaceutical company and the FDA with comparative information about the relative safety and effectiveness of the new drug. If the data is strong, the FDA may grant accelerated approval based on Phase II results. Accelerated approval allows patients access to the drug while the larger phase III study is underway. If the phase III study is negative, the FDA approval will be withdrawn.
Phase III studies are typically randomized studies with hundreds or thousands of patients that can last many years. In the end, the information provides the pharmaceutical company and the FDA with a more thorough understanding of the effectiveness of the drug, the benefits, and the range of possible adverse reactions.
Expanded Access clinical trials allow a patient to gain access to an investigational product when no comparable or satisfactory alternative therapy options are available and enrollment in a clinical trial is not possible. For drugs nearing FDA approval, a formal Expanded Access clinical trial may be available.
Expanded Access compassionate use, also known as a Single Patient Expanded Access, is a pathway by which a physician can request permission from the FDA to use an investigational product for a single patient when no comparable or satisfactory alternative treatment options are available. Your oncologist will need to request the drug on your behalf directly from the pharmaceutical company. If the company agrees, then the FDA will need to approve the use.
Phase IV studies, often called post-marketing surveillance trials, are conducted after a drug has been FDA-approved for marketing. Pharmaceutical companies have several objectives at this stage: (1) to compare a drug with other drugs already in the market; (2) to monitor a drug’s long-term effectiveness and impact on a patient’s quality of life; and (3) to determine the cost-effectiveness of a drug therapy relative to other traditional and new therapies.